
Familial hypercholesterolemia is the most common genetic disease characterised by increased LDL cholesterol from birth.
This pathology causes an increase in plasma cholesterol concentrations, mainly that transported by low-density lipoproteins (LDL-C).
It is estimated that it affects some 200,000 people in Spain , some 3 million in Europe and 34 million worldwide.
This means that it is a very common disorder since at least 1 in 250 people in the general population suffers from it.
In addition, it is the most common cause of premature cardiovascular disease. Pedro Mata, president of the Familial Hypercholesterolemia Foundation, states, “people with untreated familial hypercholesterolemia have a risk up to 20 times higher of presenting premature cardiovascular disease.”
And is that cardiovascular disease manifests itself in more than 50% of patients with familial hypercholesterolemia before 55 years of age. In addition, those who suffer from it have a high risk of presenting a myocardial infarction or other vascular atherosclerotic diseases at an early age.
Causes
This disease is caused by a mutation in the gene that codes for the LDL receptor (rLDL), which is responsible for removing cholesterol from the blood in the liver.
As explained by the Family Hypercholesterolemia Foundation, by having fewer receptors, either partial or total, LDL cholesterol increases considerably in the blood, favouring its deposit in the arteries and the development of a plaque that can narrow the lumen of the arteries, leading to atherosclerosis.
In short, “people affected by this disease have high cholesterol levels because cholesterol is not eliminated correctly in the liver due to a lack of receptors .”
Symptoms
Some indications of familial hypercholesterolemia, according to the Familial Hypercholesterolemia Foundation, are:
- Very high LDL cholesterol.
- History of myocardial infarction or atherothrombotic diseases in the family at an early age.
- Presence of other symptoms, such as arcus corneae and tendon xanthomas.
Prevention
Being a genetic disease, it cannot be prevented, but it can be controlled thanks to existing treatments that must be administered for life since it is a chronic disease.
types
According to the Familial Hypercholesterolemia Foundation, familial hypercholesterolemia can be of two types, depending on its transmission mechanism:
Heterozygous: One of the alleles has a mutation in the gene, and the other is normal. In this case, the patient has 50% of the complement of normally functioning LDL-receptors; the rest are absent (null allele mutations) or do not function properly (defective allele mutations).
Homozygous: Both alleles are defective (from the father and mother), resulting in a virtually complete absence of LDL receptors. It is a rare form of familial hypercholesterolemia that occurs when the same mutation in the LDL-r gene is inherited from both parents. It is estimated that it affects one case per million people. However, it is higher in certain regions or countries, presumably due to a founder effect and the isolation of a population.
It is important to point out that a person affected by this disease has a 50% chance of transmitting the abnormal gene to their descendants, sons and daughters, and a 50% chance of passing on the correct genetic information. Therefore, about half of the members of a family will inherit hypercholesterolemia.
Diagnosis
As mentioned before, a person affected by this disease has a 50% chance of transmitting the abnormal gene to their descendants. Therefore, if a child or adult, the son of a patient with familial hypercholesterolemia, has normal cholesterol levels, it is very likely that she has inherited the normal gene and therefore will not develop the disease or transmit it to her offspring. However, some studies have shown that there may be up to 8% of people carrying a mutation with normal cholesterol levels. In this case, they can pass on the defective gene to their offspring. Therefore, it is very important to carry out a genetic diagnosis.
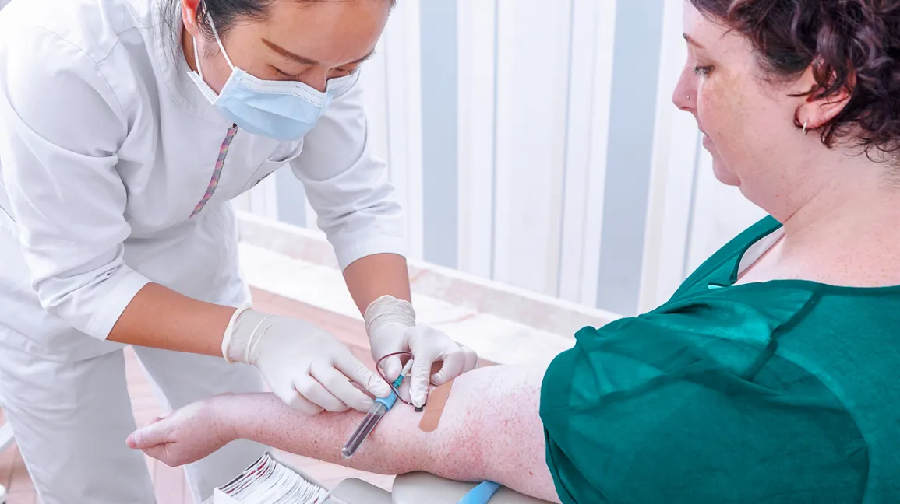
The diagnosis is very simple, “Just like the heel prick test, abnormal cholesterol levels can be detected by dry chemistry.
It is a “very simple and effective” test, but one that is not currently performed in hospitals. Hence, there is a very high underdiagnosis of the disease. According to data from the expert, “more than 80% of patients with familial hypercholesterolemia would be undiagnosed”.
The clinical diagnosis is based on elevated LDL-C concentrations, a family history of hypercholesterolemia, premature coronary artery disease, and the presence of xanthomas and arcus corneae.
According to the consensus document Diagnosis and treatment of familial hypercholesterolemia in Spain, “the diagnosis can be suspected in the presence of LDL levels > 190 mg/dl or with LDL > 150 mg/dl when there is genetic confirmation . of hypercholesterolemia or at least evidence of vertical transmission of hypercholesterolemia and premature coronary disease in one of the parents. “It has been shown that LDL-C levels discriminate well between those children with and without hypercholesterolemia before 10 years”.
For diagnosis, “it is recommended to obtain the mean of 2 determinations of the lipid profile at least two months apart due to biological variability in childhood and to rule out the causes of secondary hypercholesterolemia in childhood-adolescence”.
There is no single criterion regarding the age at which the diagnosis of hypercholesterolemia should be made. In general, “diagnosis is recommended between 2 and 10 years”.
The diagnosis of the homozygote should be made around two years of age or even earlier and is based on: plasma concentration of LDL-C without treatment > 500 mg/dl or LDL-C with treatment > 300 mg/dl, presence of xanthomas before ten years of age and history of hypercholesterolemia or genetic diagnosis in both parents. Both parents should have hypercholesterolemia and be obligate heterozygotes for the same mutation causing familial hypercholesterolemia.
Pedro Mata stated, “for every six people diagnosed with familial hypercholesterolemia and treated, a coronary episode would be avoided in 10 years”, hence the importance of its early detection.
Treatments
According to the consensus document Diagnosis and treatment of familial hypercholesterolemia in Spain, all adults with familial hypercholesterolemia should be treated with dietary measures and lipid-lowering drugs from the moment of diagnosis.
Several studies have shown that intensive lipid-lowering treatment has beneficial effects in reducing carotid intima-media thickness and improving endothelial function. Furthermore, observational studies have confirmed the cardiovascular benefit of statins in patients with familial hypercholesterolemia.
The basis of treatment is using a potent statin such as atorvastatin or rosuvastatin in monotherapy or in combination with ezetimibe and resins if the therapeutic objective is not achieved. Experts recommend that most of these patients be followed up in specialised centres in primary care and complex cases.
There is agreement that LDL-C treatment targets in children need not be as low as in adults, and there is no evidence for an absolute or relative target. This panel recommends a plasma LDL-C of <130 mg/dl from 14 years of age and <160 mg/dl in those under 14 years of age, except if there is another cardiovascular risk factor or a history of very premature coronary heart disease in the affected parent, where targets can be more stringent.
Diet is the basis of treating familial hypercholesterolemia in children and adolescents, reducing LDL-C by up to 15%. An adequate supply of energy and nutrients is essential to maintain adequate growth and body weight. In addition to a correct diet, physical activity should be promoted, not smoking.
Statins are safe and effective in children. Any statin approved by regulatory agencies can be used with dose titration based on clinical response and age. Adding resins or ezetimibe should be considered if LDL-C targets are not achieved with statins. Statins are contraindicated in pregnancy, so adolescent girls should be warned.




{kind=link}